Applications
- NAMD: Molecular Dynamics
- ChaNGa: N-Body Cosmological Simulations
- Episimdemics: Contagion in Social Networks
- OpenAtom: Ab initio Molecular Dynamics
- CSE: Computational Science and Engineering Applications
- PSTIP: Parallel Stochastic Integer Programming
Molecular Dynamics - NAMD
NAMD, recipient of a 2002 Gordon Bell Award, is a parallel molecular dynamics application designed for high-performance simulation of large biomolecular systems. NAMD is a result of many years of collaboration between Prof. Kale, Prof. Robert D. Skeel, and Prof. Klaus J. Schulten at the Theoretical and Computational Biophysics Group (TCBG) of Beckman Institute.

NAMD is distributed free of charge and includes source code. Charm++, developed by Prof. Kale and co-workers, simplifies parallel programming and provides automatic load balancing, which was crucial to the performance of NAMD. It is used by tens of thousands of biophysical researchers with production versions installed on most supercomputing platforms. NAMD scales to hundreds of cores for small simulations and beyond 300,000 cores for the largest simulations. See the TCBG NAMD web site for NIH supported download and tutorial instructions.
The dynamic components of NAMD are implemented in the Charm++ parallel language. It is composed of collections of C++ objects, which communicate by remotely invoking methods on other objects. This supports the multi-partition decompositions in NAMD. Also data-driven execution adaptively overlaps communication and computation. Finally, NAMD benefits from Charm++'s load balancing framework to achieve unsurpassed parallel performance. See PPL NAMD research page for more details.
N-Body Cosmological Simulations - ChaNGa

ChaNGa (Charm++ N-Body Gravity Simulator) is a cosmological simulator to study formation of galaxies and other large scale structures in the Universe. It is a result of interdisciplinary collaboration between Prof. Kale, Prof. Thomas Quinn of University of Washington and Prof. Orion Lawlor of University of Alaska Fairbanks. ChaNGa is a production code with the features required for accurate simulation, including canonical, comoving coordinates with a symplectic integrator to efficiently handle cosmological dynamics, individual and adaptive time steps, periodic boundary conditions using Ewald summation, and Smooth Particle Hydrodynamics (SPH) for adiabatic gas.
ChaNGa implements the well-known Barnes-Hut algorithm, which has N log N computational complexity, organizing the particles involved in the simulation into a tree based on Oct, Orthogonal Recursive Bisection (ORB), or SFC decompositions. In order to compute the pair interaction between particles and collections of particles on different processors, parts of the tree needed for the computation are imported from the remote processors which own them. ChaNGa uses the Charm++ Salsa parallel visualization and analysis tool. Visualization in the context of cosmology involves a large amount of data, possibly spread over multiple processors.
ChaNGa has been scaled to 32K cores, and has been ported to GPU clusters. Over time, ChaNGa is being actively developed and improved, with an eye for efficient utilization and scaling of current and future supercomputing systems. See ChaNGa web site for download and instructions.
Contagion in Social Networks - Episimdemics
The study of contagion effects in extremely large social networks, such as the spread of disease pathogens through a population, is critical to many areas of our world. Scaling Agent-based Simulation of Contagion Diffusion over Dynamic Networks on Petascale Machines Applications that model dynamical systems involve large scale, irregular graph processing. These applications are difficult to scale due to the evolutionary nature of their workload, irregular communication and load imbalance. EpiSimdemics implements a graph based system that captures dynamics among co-evolving entities, while simulating contagious diffusion in extremely large and realistic social contact networks. EpiSimdemics relies on individual-based models, thus allowing studies in great detail. This paper presents a novel implementation of EpiSimdemics in Charm++, which enables future research by social, biological and computational scientists at unprecedented data and system scales. We present new methods for application-specific decomposition of graph data and predictive dynamic load migration and demonstrate the effectiveness of these methods on Cray XE6/XK7 and IBM Blue Gene/Q.
Ab initio Molecular Dynamics - OpenAtom

Many important problems in material science, chemistry, solid-state physics, and biophysics require a modeling approach based on fundamental quantum mechanical principles. A particular approach that has proven to be relatively efficient and useful is Car-Parrinello ab initio molecular dynamics (CPAIMD). Parallelization of this approach beyond a few hundred processors is challenging, due to the complex dependencies among various subcomputations, which lead to complex communication optimization and load balancing problems. We have parallelized CPAIMD using Charm++. The computation is modeled using a large number of virtual processors, which are mapped flexibly to available processors with assistance from the Charm++ runtime system. See the OpenAtom web site for more details.
Computational Science and Engineering Applications - CSE
Professor P. Geubelle and S. Breitenfeld of the Computational Solid Mechanics Group have developed CrackProp, an explicit Finite Element method simulation of viscoelastic and plasto-elastic mechanics. The simulation tracks conventional volumetric elements, coupled by flat "cohesive" interface elements using the Charm++ Finite Element Framework.

Professor J. Dantzig and Jun-Ho Jeong of the Solidification Processing Lab have parallelized their dendritic growth metal solidification application using the Charm++ Finite Element Framework. This adaptive mesh, implicit solver fluid dynamics application is quite different from the explicit structures codes normally used.
The framework handles the changes to the adaptive mesh by re-assembling the parallel mesh, repartitioning, and redistributing the mesh pieces. Since the mesh changes only rarely, this does not significantly impact the speed of the simulation. The implicit solvers are implemented using the conjugate gradient method, which solves a global matrix using local operations.
Advanced Rocket Simulation
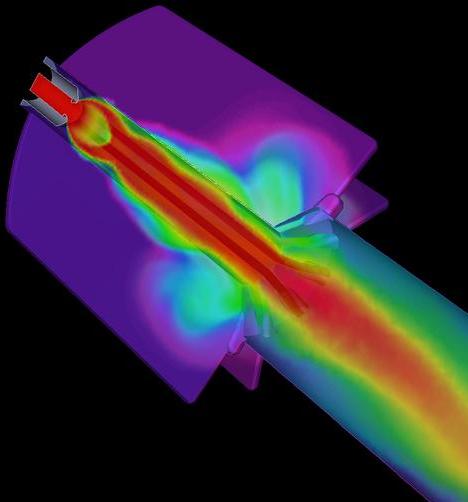
The principal goal of Center for Simulation of Advanced Rockets (CSAR) is detailed, integrated, whole-system simulation of solid propellant rockets under both normal and abnormal operating conditions. Various components of the integrated rocket simulation codes, ROCFLO, ROCSOLID, and ROCFACE have been ported to Adaptive MPI with minimal changes and with little effort, providing them with automatic load balancing capabilities. The AMPI implementation also enhances the modularity of rocket codes by using separate sets of virtual processors for ROCFLO and ROCSOLID. Based on a similar programming paradigm, a prototype finite element method (FEM) framework has been developed. Although originally designed for finite element applications, it is general enough for unstructured mesh applications. Using the FEM framework, code developers write only sequential code that implements the node and element-specific computations of the FEM solver, while the details of parallelism such as partitioning, communication, and load balancing are taken care of by the framework.
PSTIP - Parallel Stochastic Integer Programming

Stochastic optimization is used for optimal resource allocation under uncertainty. Application of stochastic programming spans a diverse set of fields ranging from production, financial modeling, transportation (road as well as air), supply chain and scheduling to environmental and pollution control, telecommunications and electricity. When integer optimization is required, then branch-and-bound are accepted norms.
Unlike typical, iterative scientific applications, this application has some very interesting characteristics that make it challenging to realize a scalable design. The total amount of computation required to find optima is not constant across multiple runs. This challenges traditional thinking about scalability and parallel efficiency. It also implies that reducing idle time does not imply quicker runs. The sequential grains of computation are quite coarse. They display a wide variation and unpredictability in sizes. The structure of the branch-and-bound search tree is sensitive to several factors, any of which can significantly alter the search tree causing longer times to solution.
Our parallel design exploits simultaneous parallelism on two different fronts - parallel exploration of the branch-and-bound tree vertices tree and parallel evaluation of scenarios. Our designs show strong scaling to hundreds of cores. See the report for more details.